September 18, 2013 — This article discusses the history of the STERIS System 1 Liquid Chemical Processor and its de facto recall (by the Food and Drug Administration).
Click here to learn about a program that Dr. Muscarella designed for healthcare facilities to reduce the risk of infection-control breaches and associated infections.
Further, this article is based on a more complete article. Email Dr. Muscarella at Larry@LFM-HCS.com to receive a copy. In addition, it is suggested that this article be read along with two other articles that Dr. Muscarella also wrote and that are available by clicking here (article #1) and by clicking here (article #2), respectively.
Introduction
Few violations of infection-control standards have received more scrutiny than recent reports of reprocessing breaches at three medical centers within the Veterans Health Administration (VHA).(1-5)
Located in Murfreesboro (TN), Miami (FL) and Augusta (GA), these three VA medical centers this past winter did not properly clean, disinfect, or use reusable endoscopic instrumentation, potentially exposing more than 10,000 U.S. veterans to HIV, the hepatitis C virus and other infectious agents.(1-5)
As the incidents at these three medical centers and others demonstrate, the improper reprocessing of reusable instrumentation can be associated with adverse outcomes.
[Are you in the market to buy a steam autoclave or a steam sterilizer? Click the link.]
The STERIS System 1 Sterile Processing System (“System 1”)—an automated reprocessing device that was introduced to the U.S. market in 1988—was labeled both to achieve liquid chemical sterilization using peracetic acid and to produce “sterile” rinse water using a 0.2 micron bacterial filter.(6-11)
This article is a shortened version of a more complete article, in PDF format, that can be downloaded in its entirety by clicking here. This more complete PDF article includes several box articles that are discussed, below, but that are not included in this article.
Save the System 1, no other device has been labeled with either of these two claims.(12) The System 1 was labeled to “sterilize” not only flexible endoscopes and other reusable endoscopic instrumentation, but also laparoscopes and arthroscopes; microsurgical, ophthalmic, and dental instruments; and biopsy forceps, among other types of surgical instruments.(6-11)
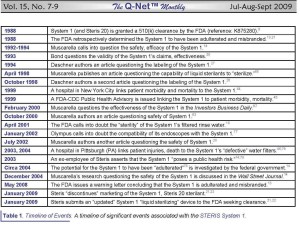
The reader is directed to Table 1, “A Timeline of Events,” below, which is also available on p. 18S1 of the more complete article upon which this article is based. This table and page are available by clicking here.
FDA warning letter
The Food and Drug Administration (FDA) published a warning letter, dated May, 15, 2008, asserting that the System 1 is “adulterated” and “misbranded.”(13)
With potentially significant infection-control implications, this warning letter’s conclusions were the apparent culmination of a federal investigation that began circa 2004 to evaluate the safety, effectiveness, and labeling of the System 1.(14) The FDA asserted in this letter that, between both 1988 and 2009, both the System 1 and its accompanying sterilant – a single-use, peracetic acid-based concentrate known as the “STERIS 20” – have undergone several “significant changes or modifications” that could “significantly affect (their) safety or effectiveness.”(13)
In short, the FDA’s warning letter asserted that the System 1 and STERIS 20 sterilant were without a clearance or approval as required to be legally marketed,(13) adding that the agency was not notified of the System 1’s significant changes as required by the Federal Food Drug and Cosmetic Act (“FD&C Act”)(15-17)—changes that include, for example, altering the STERIS 20’s formulation.(13)
[Are you looking to buy surgical instrument trays? Click the link.]
These changes—each of which the FDA’s warning letter itemized and stated was unapproved and “itself would necessitate submission” of a new application for clearance or approval—raised doubts, according to the FDA, about, not only the STERIS 20’s chemical “stability,” but also the System 1’s safety and “ability to sterilize” surgical instruments and flexible endoscopes.(13)
The Food Drug and Cosmetic Act (“FD&C Act”)
The Medical Device Amendments to the FD&C Act (in 1976) prohibit the introduction into interstate commerce† of a significantly modified device whose changes did not receive from the FDA a “510(k) clearance” or a “premarket approval” (PMA).(15-17)
[† Interstate commerce refers to the manufacture, packaging, shipment and commercial distribution (e.g., marketing, selling, and buying) of a product, including a medical device, within and across the States’ borders. This term originates from the Commerce Clause of the U.S. Constitution (see: the U.S. Constitution’s Article 1, Section 8, Clause 3).(15-17)]
A mislabeled device?
The implications of the FDA’s warning letter were salient and complement conclusions the FDA previously published in another letter it wrote seven years earlier, in 2001, suggesting that the System 1 may be mislabeled.(18)
Specifically, this letter stated that the “association of the STERIS System 1 processor with patient infections usually caused by waterborne organisms leads (the FDA) to question the ability of the processor to provide a sterile water rinse,” adding that the FDA “believe(s) that the processor may not be functioning as it is labeled” (i.e., is mislabeled).(14,18-20)
[Are you looking to buy a used washer-disinfector? Click the link.]
STERIS’s letter to customers
STERIS responded to the FDA’s 2009 warning letter in a letter it wrote to its customers, dated January 20, 2009 (see: Table 1, which is inserted into this article, above).(21)
Aiming to market another “sterilizing” device, STERIS stated in this letter that two weeks earlier (in early January) it had applied for a new 510(k) clearance to market this modified model of the System 1—known as “the System 1E.”(21,22)
Attention: Read more about the STERIS System 1E in a related article by Dr. Muscarella entitled “The STERIS System 1E Liquid Chemical Sterilant Processing System: Looking Back and Forward” — click here to read it. (Also refer to the FDA’s discussion of the System 1E by clicking here.)
According to STERIS, it sought to market this updated device to achieve “liquid chemical sterilization” and to produce “sterile” filtered rinse water from a tap.(21,22)
While the FDA has published its definition of “liquid chemical sterilization” here, Dr. Muscarella has raised some scientific questions about this definition. Click here to read his article that details some of these concerns.
STERIS further stated in this letter that it was “discontinuing” sales in the U.S. of the modified (and unapproved) System 1.(21) But the details of this action were qualified, if not unique.
As it described in this letter and a contemporaneously published press release, STERIS would continue to sell in the U.S the modified (and unapproved) System 1 for at least two more years (albeit only as a “product replacement”—that is, to replace a previously purchased System 1 processor).”(21,22)
Discussion
A study of the STERIS System 1’s history, regulatory oversight, and marketing teaches a litany of insightful, if not fascinating and far-reaching, lessons about medical devices and infection control.
Indeed, articles focusing on risk management and current instrument-reprocessing practices would be arguably incomplete if they were not to discuss the System 1’s recent censure and continued use by, among others, VA medical centers—notwithstanding the FDA having concluded in 2009 that the System 1 was adulterated, misbranded, and without regulatory approval(13) (considerations that would ordinarily result in prompt termination of a device’s use).
[Are you in the market to buy an instrument detergent by STERIS or by another company? Click the links.]
Providing a rare glimpse into the marketing and use of infection-control devices, a study of the STERIS System 1 also yields insight into:
- the mutual quest by manufacturers and healthcare practitioners for the ideal, rapid-acting processor to “sterilize” heat–sensitive surgical instruments;
- the complex financial and “working” relationships between manufacturers and healthcare organizations and institutes;23,24 and
- the acquiescence through the years by healthcare organizations of the System 1’s seemingly implausible(14,25-28) “guarantee”(14,26,29) both: (i) to achieve “liquid chemical sterilization”;(6-11) and (ii) to produce “sterile” filtered rinse water from a tap.(6,8,9,14)
(“Sterility” cannot be “guaranteed”(26) but is defined as a probability for contamination—namely as a “sterility assurance level,” or SAL, of, for example, 10^-6.(73))
Adding to the STERIS System 1’s mystique and alluring singularity, these two labeling claims— that this device achieves “liquid chemical sterilization” and produces “sterile” filtered rinse water from a tap have proved to be elusive for other manufacturers.
A. The Food Drug and Cosmetic Act (“FD&C Act”)
Legitimate questions may arise within the healthcare community about the STERIS System 1 and the soundness of its continued use (and sale, whether or not as a replacement) from 2009 through 2012, when this device could no longer be legally used in the U.S.
These questions follow both from the FDA’s warning letter, which concluded that the System 1 (and STERIS 20) had been adulterated and misbranded since 1988,(13,21) and from a basic understanding of the FD&C Act, which prohibits the manufacture; commercial distribution; receipt; and introduction into interstate commerce of an adulterated and misbranded device.(16,17)
[Are you looking to buy an enzymatic detergent to clean surgical instruments? Click the link.]
Indeed, some provisions of the FD&C Act also oversee a device’s clinical use. A significantly modified class II or class III device without a 510(k) clearance or PMA (but that would otherwise require either) may be referred to as “investigational,” requiring for its use the approval of an investigational device exemption” or IDE.(13,17)
According to the FDA, in addition to being misbranded for having been introduced into commercial distribution without a 510(k) clearance, the System 1 and STERIS 20 are also adulterated for lacking a PMA or an approved IDE.13 Among other provisions, investigational devices (i.e., “unapproved devices”(17)) notably require informed patient consent.
(Refer to: the box article: “What is an ‘investigational’ device?,” which is provided on p. 18S2 of the article that can be read in the more complete, PDF version of this article, which may be downloaded by clicking here.)
Nevertheless, STERIS claimed that, in addition to selling in the U.S. both the System 1 and STERIS 20 for at least two more years (albeit conditionally), medical centers including those within the VHA could continue using the System 1 and STERIS 20, “without any change” in clinical practice (and without notifying the patient).(21,22)
In the context of the FD&C Act’s provisions regarding investigational devices—which, like the System 1, are without a 510(k) clearance or PMA (and are not otherwise exempted)(13,15,17)—this instruction is understandably confusing and controversial.
B. Mission statements
The mission statements of healthcare organizations in the fields of infection control, aseptic technique, and instrument reprocessing typically pledge a commitment to advancing public health, prioritizing patient safety, and preventing healthcare-associated infections.
As a public display of their avowed responsibilities, these organizations, in addition to publishing guidelines, may periodically issue position statements, sentinel event alerts, or advisories, providing an important and worthy system of “checks and balances” that serves to monitor the safety and effectiveness of medical devices used by their respective membership.(30-34)
A review of the literature finds that these organizations … [continued. This and some other paragraphs in this section are continued in the more comprehensive article upon which this article is based. Click here to read it.]
[Are you in the market to buy Cidex OPA, Metricide (14-day), Rapicide, Metricide OPA, Resert XL, or Cidex (14-day) to high-level disinfect your instruments? Click the links.]
C. The Abtox Plazlyte System
Providing insight and perspective, the FDA in 1998 censured the Abtox Plazlyte Sterilization System (“Plazlyte System”), which, like the Steris System 1, was marketed to “sterilize” heat-sensitive surgical instruments using peracetic acid (as a low-temperature vapor mixed with other chemicals).(53)
Cleared by the FDA in 1994 and first discussed by Dr. Muscarella more than a decade ago, the Plazlyte System was investigated in 1998 for its association with six patient injuries (no deaths) following ophthalmic surgery.(53) As a consequence, the FDA issued an alert that year expressing concerns about the Plazlyte System’s safety.(54)
A comparison of the Abtox Plazlyte’s marketing and labeling claims to those of the Steris System 1’s (and Steris 20’s) yields some interesting similarities.
In addition to both “sterilizing” devices having been linked to patient injury,(14,20,44-46,53,54) the Plazlyte System and System 1 share other features, too, some of which are listed in Table 3 on p. 18S2 (which can be read by downloading the more complete PDF version of this article at this link).
Like the System 1, the marketed model of the Plazlyte System reportedly was significantly different from the model that the FDA had originally cleared—for example, like the reformulation of the Steris 20,(13) the gas ratio of the sterilant used by the Plazlyte System had been modified without clearance or approval by the FDA.(53-56)
Nevertheless, despite their similarities, there are differences, too. Whereas the Plazlyte System was promptly removed from the market on March 31, 1998,(53-56) the STERIS System 1, according to its manufacturer, will continue to be sold, along with the STERIS 20, for at least two more years (albeit conditionally).(21,22)
Notably, the FDA has not, to date, discussed whether the continued use of the System 1 is safe, appropriate, and, as its manufacturer asserts, neither warrants changes in clinical practice nor the notification of doctors or patients.(22)
Click here to read Dr. Muscarella’s related article that discusses the AbTox Plazlyte System in the context of the eye disorder “TASS” (toxic anterior segment syndrome) and the sterilization of ophthalmic instruments.
D. Surgical instrument manufacturers
Some manufacturers of reusable surgical instruments discuss the Steris System 1 in their labeling. For example, the operator’s manuals and reprocessing instructions provided by some rigid-endoscope manufacturers claim that the Steris System 1 is compatible with, and has been validated for the “sterilization” of, their endoscopes.(57-61)
But, apparently presenting another breakdown in an important system of checks and balances, a review of the literature finds that the requisite validation and verification data to support these claims about the System 1 are lacking.
Further, as a consequence of the conclusions of the FDA’s warning letter(13) (and Steris’s reply to it(21,22)), these operator’s manuals and other labeling referencing the System 1 would presumably warrant revision.
[Are you looking to buy a used colonoscope? Click the link.]
E. Dr. Muscarella’s perspectives
The 510(k) clearance of which was first queried by Bond in 1993,(28) few researchers have questioned the safety, effectiveness, and labeling claims of the STERIS System 1.(14,20,25-27) Published reports,(6-10) guidelines(47-50) and evaluations(14,24,51) (and surgical-instrument operator’s manuals(57-61)) discussing the System 1 often overlook its shortcomings—for example, not acknowledging that the validation and verification data in support of the System 1’s claim to achieve “sterilization,” under worst-case clinical conditions, are lacking.
Nor do these publications note that the STERIS 20’s peracetic acid has not been challenged (as otherwise required of liquid sterilants) by the Association of Official Analytical Chemists’ (AOAC) Sporicidal Test(28)—a standardized test otherwise required by the Environmental Protection Agency (EPA) to evaluate the effectiveness of a sterilizing agent.(62)
Instead of questioning the System 1’s singular claims, these publications focus on this device’s apparent strengths, of which there are some, including its ease-of-use; relatively rapid-acting cycle; containment and use of a single-use disinfectant; portability; and its relatively small footprint.(6-10)
During the past two decades, Muscarella has authored several articles published in medical journals that discuss the inherent limitations of liquid sterilants.(20,25,63-65)
In several of his articles, Muscarella discusses that, while some liquid chemical sterilants, such as peracetic acid, may under certain conditions be sporicidal, their limitations—including that their potentially toxic residues be removed from the instrument’s surfaces following chemical immersion using large volumes of rinse water, the microbial quality of which is not routinely monitored microbiologically and, therefore, rarely known—preclude claiming that any processor can reliably and consistently achieve liquid sterilization, particularly of instruments, such as flexible endoscopes, that may feature inaccessible surfaces.(25-27,65-69)
Further, in agreement with Daschner,(26) Muscarella has also written not only that the “guarantee” that a process achieves “sterilization” is dubious, but also that the limitations of 0.2 (and 0.1) micron bacterial water filters, used by virtually every automated endoscope processor to improve the quality of its rinse water, prevent these filters from reliably producing, from a facility’s tap, “sterile” water (or any quality of water that exceeds that which is produced by reverse osmosis)—a shortcoming of bacterial filters that is an Achilles’ heel and belies any liquid-based processor’s claim to achieve “sterilization” of surgical instruments.(19,20,25,52,63,65,68,70)
In short, Muscarella has questioned on the front page of The Wall Street Journal (14) and another national daily newspaper(67) the soundness and appropriateness of labeling any automated liquid-based processor:
- to achieve liquid sterilization;
- to produce sterile filtered rinse water; and
- not to require terminal drying of the endoscopes, an otherwise requisite reprocessing step crucial to patient safety.(14,20,52)
Daschner, too, has challenged the System 1’s “sterilization” claim, referring to the System 1 instead as a device that “disinfects.”(26,27)
Like Bond, Muscarella has also questioned the scientific basis for labeling a biological indicator to monitor the effectiveness of this or any other liquid-based “sterilization” process.(14,28,66)
Attention: Read Dr. Muscarella’s related article “The STERIS System 1E Liquid Chemical Sterilant Processing System: Looking Back and Forward” (click here), which discusses the use spore strip tests to monitor liquid cehcial sterilizaiotn process. AS Dr. Muscarella discusses, the FDA distinguishes a spore strip test from a biological indicator, or BI.
Though some have maintained the validity of the STERIS System 1’s claims,(6-11,14,29,47-51,57,71,72) the conclusions of the FDA’s warning letter, along with the manufacturer’s discontinuation of the System 1 and STERIS 20 (albeit with some qualifications), is consistent with and reaffirms the merit of several of the concerns about the System 1’s safety and labeling that Muscarella, Bond and Daschner have published for years.
That the Steris System 1 was recently declared by the FDA to be adulterated and misbranded since 1988 is consistent with and reaffirms the concerns that Muscarella, Bond and Daschner had previously published about this device’s safety and labeling.
Conclusions
An infection-control device whose use had been common in the U.S. up until circa 2009 (this article was written in January, 2013),(14,48) the article upon which this article is based was published in 2009 and the first to discuss the censure and discontinuation of the STERIS System 1 as they may apply to the FD&C Act.
Indeed, a review of this Act’s relevant provisions—along with the FDA’s warning letter; both the medical and legal literature; and articles discussing the marketing, design changes, and recall of the Abtox Plazlyte System—provides important insight and perspective, suggesting that the approbation or endorsement of the use of any adulterated and misbranded device would appear to be injudicious and incongruous with patient safety.
[Are you looking to buy a washer-sterilizer or disinfector? Click the link.]
Several questions remain unanswered, such as whether the use of an adulterated or misbranded device might adversely affect a medical facility’s accreditation, or whether in past years the use of another adulterated or misbranded device had been sanctioned or approved.
While patient injuries linked to the use of an unapproved device (without an IDE) would seemingly be prejudicial, less clear is whether it is necessary to notify patients whose injuries were linked to a device subsequently determined by the FDA to have been adulterated and misbranded at the time of the injuries.(44-46)
To be sure, several lessons about infection control and instrument reprocessing are taught by this study of the STERIS System 1 and STERIS 20, underscoring the importance to patient safety:
- of healthcare staff members having a basic understanding of, first, the inherent limitations of liquid sterilants and 0.2 micron bacterial water filters; and, second, the regulation of medical devices; along with, third, consideration of all of the potential implications, if not risks, associated with using an adulterated, misbranded, or otherwise unapproved device;
- of medical device manufacturers being circumspect and not incautiously claiming in their operator’s manuals that a specific device “sterilizes” their surgical instruments, unless these manufacturers have in their quality-assurance records validation and verification data substantiating the claim; and
- of healthcare organizations enhancing their commitment:to patient safety; to their respective mission statement’s pledges; to the issuance of timely safety alerts; and to writing guidelines that are evidence-based and revised as warranted.
Also taught by this study’s lessons is the importance of the both FDA and the FD&C Act to public health. Indeed, this study of the STERIS System 1 teaches many lessons about infection control, instrument reprocessing, and patient safety. No time is better than now to learn them. [The End.]
References to this article may be read by clicking here.
Article by: Lawrence F. Muscarella PhD posted on 1-9-2013; updated 11-20-2014. Copyright. LFM Healthcare Solutions, LLC. All rights reserved.