December 8, 2017 (2:46 pm) — A duodenoscope has been linked, possibly for the first time, to the “probable transmission” of a uniquely resistant type of superbug, I have learned.
This incident is disclosed in an otherwise unknown regulatory report that the duodenoscope’s manufacturer submitted to the FDA earlier this year, in August.
Seven months earlier, in January, the FDA had informed hospitals about a “design issue” with the duodenoscope model described in this regulatory report, according to the agency, “that could increase the risk of patient infection” during ERCP.
In July, the manufacturer recalled this device.
This August regulatory report describes a patient who was infected (or colonized) with colistin-resistant Klebsiella pneumoniae following endoscopic retrograde cholangiopancreatography performed apparently earlier in the spring. The same duodenoscope had been used nine days earlier on a patient already infected with this superbug, according to the report.
Testing confirmed that the superbug described in this report was resistant to cefotaxime, a third-generation cephalosporin antibiotic. This CTX-resistant Kl. pneumoniae strain was also found to carry a unique gene — the mcr-1 gene — which confers the bacteria’s resistance to colistin, a “last resort” antibiotic.
Whether these bacteria were tested and confirmed to be CRE due to a demonstrated resistance to carbapenems, too, is not discussed in the report. Colistin-resistant bacteria pose a serious and emerging threat to public health, both in the U.S. and globally.
This article herein is apparently the first to publicly disclose this case of possible transmission of colistin-resistant Enterobacteriaceae bacteria during ERCP. These nightmarish bacteria are virtually untreatable.
The manufacturer’s August regulatory report may be read in its entirety by clicking here. (This is one of six reports this manufacturer filed with the FDA documenting this mcr-1 incident.)
![]() Consulting Services: LFM-Healthcare Solutions, LLC provides medical expertise for hospitals, manufacturers and the public, specializing in healthcare-associated infections linked to medical equipment. Services include litigation support. Years of experience. Reasonable rates.
Consulting Services: LFM-Healthcare Solutions, LLC provides medical expertise for hospitals, manufacturers and the public, specializing in healthcare-associated infections linked to medical equipment. Services include litigation support. Years of experience. Reasonable rates.
The first time?
This August regulatory report, filed by Pentax Medical (of Montvale, NJ) who manufactured the duodenoscope, may document, for the first time, the possible patient-to-patient transmission during ERCP of a superbug containing the mobilized colistin resistance (mcr-1) gene. (Insufficient data are currently available to conclude that the device caused the infection. Nor does “possible” transmission of a superbug confirm actual transmission.)
In general, bacterial strains that previously caused infections linked to a duodenoscope, while resistant to carbapenem antibiotics, were not pandrug-resistant (PDR) and generally were susceptible to colistin (as described in here).
Providing evidence of the potential clinical significance of this case, I could not identify another documented instance in the FDA’s medical device database, or in the published medical literature, linking a duodenoscope (or any type of endoscope) to an infection caused by a superbug carrying the mcr-1 gene.
This device database is sometimes referred to as the FDA’s MAUDE database.
(A 2017 report discusses a hospital in Turkey that identified an outbreak of colistin-resistant Pseudomonas aeruginosa in 2013 reportedly due to a contaminated bronchoscope, but not a duodenoscope.)
As many as five patients may have been exposed to the duodenoscope, according to the August regulatory report. As is the FDA’s general practice, this regulatory report does not disclose the name of the hospital where the possible transmission of this superbug with the mcr-1 gene occurred.
This August regulatory report discloses that the U.S. Centers for Disease Control and Prevention (CDC), which began investigating this case last summer, informed a manufacturer representative “that their preliminary testing results indicate that the isolates from the patients and the device appear related …”
Few other details about this incident are known. This August regulatory report does not disclose whether one or more infection-control breaches, such as the improper cleaning of the duodenoscope, had been identified.
Nor does the report reveal how the duodenoscope was reprocessed. The FDA has published a table listing those automated endoscope reprocessor (AER) models that the agency reports have been validated for reprocessing duodenoscopes.
In 2013, this Pentax duodenoscope model, with a sealed (elevator-channel) design, was publicly linked, for the first time in the U.S., to an outbreak of CRE, near Chicago (IL). This CRE strain carried the New Delhi metallo-β-lactamase — or NDM-1 — gene, however, not the mcr-1 gene. At least in the past, NDM-1 producing CRE were often susceptible to the colistin antibiotic.
CRE is an acronym for carbapenem-resistant Enterobacteriaceae.
The previous year, a hospital in the Netherlands linked an outbreak of VIM-2-producing P. aeruginosa to a different manufacturer’s duodenoscope model that similarly featured a sealed design. The outbreak’s multidrug-resistant (MDR) superbug was reportedly susceptible only to colistin.
In general, whereas these newer duodenoscope models feature an elevator channel that is intended to be “sealed” obviating reprocessing, this channel in older duodenoscope models instead is “open” requiring cleaning and disinfection.
FDA safety alert — January 2017
Last January, seven months prior to the filing of this August regulatory report, the FDA issued a safety communication entitled, “UPDATE: Importance of Following Validated Reprocessing Instructions for PENTAX ED-3490TK Video Duodenoscopes.”
In this alert dated January 17, 2017, the FDA advised hospitals, using a “blue box” format [see inserted image, below], about a “design issue” with this duodenoscope model that “could increase the risk of patient infection.”
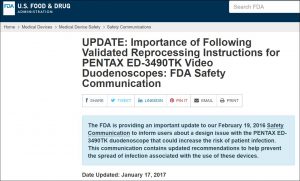
In particular, this federal alert stated that, “Cracks and gaps in the adhesive that seals the device’s distal cap to its distal tip can occur, which can lead to microbial and fluid ingress. These areas can be challenging to clean and high-level disinfect and may increase the risk of infection transmission among patients.”
The FDA’s alert further noted that the manufacturer’s reprocessing instructions “when followed correctly, are intended to effectively clean and high-level disinfect the duodenoscope channels, elevator area, and exterior of the device, and should reduce the risk of device contamination.”
The potential for the complex designs of duodenoscopes to impede reprocessing, to remain contaminated (sometimes, reportedly even when manufacturer reprocessing instructions were followed correctly), and to transmit multidrug-resistant superbugs during ERCP was a central focus, not only of an earlier safety notice the FDA published in February 2015, but also of a U.S. congressional investigation and report published in January 2016.
This congressional investigation concluded, among other salient findings, that: “The failure of FDA’s current device safety reporting system to rapidly identify duodenoscope-related, antibiotic-resistant infections, including superbug infections, should serve as warning that without a comprehensive postmarket device surveillance system that supplements self-reporting from hospitals and manufacturers, future device issues are likely to go undetected for far too long and with life-threatening consequences.”
This congressional report added that this failure “serves as just one example of the fallacy of a system that is primarily reliant on hospitals and device manufacturers to self-report information to FDA.” Nevertheless, Pentax appears to have filed this August regulatory report discussing the Klebsiella strain with the mcr-1 gene in compliance with the FDA’s device reporting regulations.
Between 2012 and spring 2015, duodenoscope models with a sealed (elevator-channel) design were linked to at least 25 different instances of antibiotic-resistant infections sickening more than 200 patients worldwide, according to this 2016 congressional report.
The FDA’s January 17th alert [see inserted image, above] provided “updated recommendations to help prevent the spread of infection” associated with the use of this ED-3490TK model, including the importance of hospitals “meticulously” following this device’s reprocessing instructions.
This safety communication also stressed that hospitals pay “close attention to any physical damage” to the device, recommending that hospitals “immediately remove from service for assessment, and repair or replace any duodenoscope that shows visible signs of damage. Examples of damage may include: loose parts, damaged channel walls, kinks or bends in tubing, holes in the distal end, cracks and gaps in the adhesive that seals the device’s distal cap or other signs of wear or damage.”
The previous day, Pentax had distributed a “field correction” letter to inform affected customers about “a potential issue associated with the distal cap of the ED-3490TK.” According to this letter, dated January 16, 2017 (which also focused on the manufacturer’s ED-3270K duodenoscope model), “During manufacturing, silicone adhesive is applied to the distal tip prior to affixing the distal cap. In some instances, cracks or gaps may form in the adhesive that may be vulnerable to fluid ingress and soiling” [see inserted image, below at right].
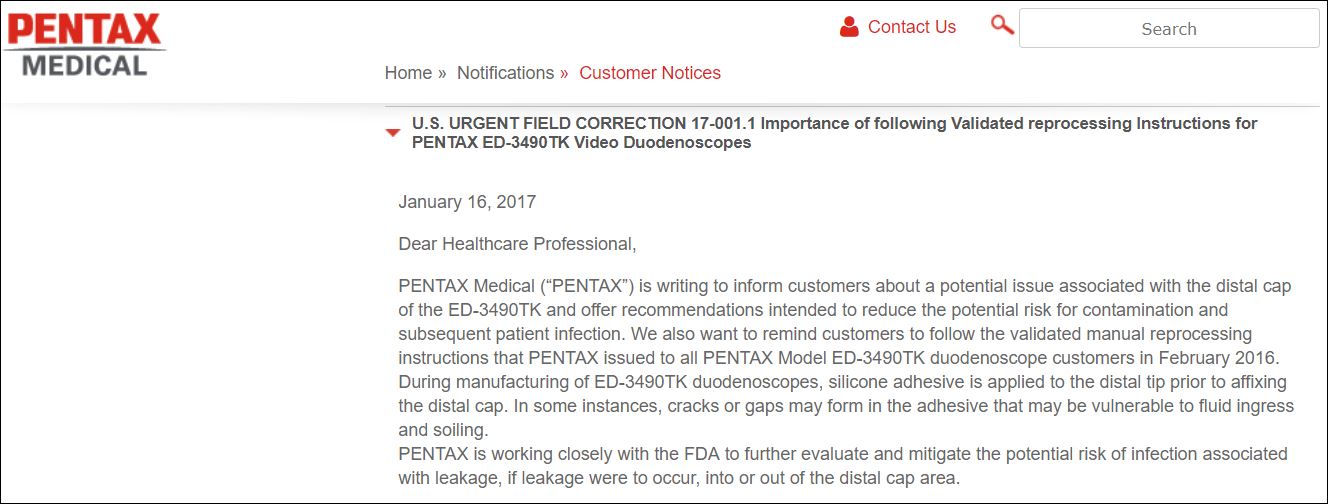
As noted in the FDA’s January 17th alert, Pentax’s field correction letter recommended that users “closely inspect their duodenoscope and remove from use (any) that show signs of physical damage.” Pentax’s letter also cautioned that “Continuing to use devices with integrity issues (i.e. holes, cracks, kinks and scratches) can contribute to persistent device contamination and subsequent patient infection.”
As another risk mitigation strategy, Pentax’s January letter stated that the company “will provide annual inspection and servicing to all customers with ED-3490TK duodenoscopes. During on-site inspections, PENTAX field staff will assess duodenoscope functionality and will send duodenoscopes for in-house servicing to recondition worn parts, as needed. PENTAX will contact your facility to schedule time to inspect your ED-3490TK duodenoscope inventory and heightened vigilance will be applied to the integrity of the distal cap during these inspections.”
Nevertheless, the FDA’s January 17th safety alert, issued the next day, did not recommend hospitals necessarily stop using the ED-3490TK model — unless, for example, the device shows visible signs of damage. (At that time in January, the FDA may not have considered the potential for a duodenoscope to transmit colistin-resistant superbugs.)
Possibly an inadvertent oversight, the FDA’s January alert also did not expressly recommend that hospitals adopt at least one of the four supplemental measures, or mitigations, that the agency had previously provided in a safety communication published almost a year-and-a-half earlier, on August 4, 2015, to reduce further the risk of CRE infections following ERCP and to increase the safety of duodenoscopes. (This alert does, however, list as a relevant footnote the safety communication discussing these four measures.)
Also in August 2015, the FDA wrote a letter to Pentax citing the company for apparent quality lapses. The same month, the two other duodenoscope manufacturers — Olympus (Center Valley, PA) and FujiFilm (Wayne, NJ) — received a similar letter from the FDA. (Please refer to the related article, “Olympus Recalls the ‘Sealed’ TJF-Q180V Duodenoscope.“)
While the ED-3490TK duodenoscope that was the subject of this January FDA safety alert (and the manufacturer’s field correction letter) is the same model identified in the August regulatory report, data verifying that this case of possible transmission of a superbug carrying the mcr-1 gene was a direct consequence of this duodenoscope’s design have not been published.
Pentax recalls the ED-3490TK duodenoscope — July 2017
Six months after the FDA issued its January 17th safety alert (and a month before the August regulatory report discussing this case of possible transmission of colistin-resistant Kl. pneumoniae during ERCP was filed), Pentax recalled the ED-3490TK duodenoscope model (and, too, the ED-3270K duodenoscope model).
In a posted recall notice dated July, 11, 2017, Pentax discussed its January 16th (2017) field correction letter, writing in this notice that “(this January) letter described the product, problem and actions to be taken. The customers were instructed to immediately remove any affected product from use; follow product labeling; (and to) ensure all reprocessing personnel are knowledgeable and thoroughly trained on the instructions of Use for manual reprocessing of the devices; clean elevator recesses and follow all reprocessing instructions; and … .”
Like the FDA’s January 17th safety alert, this posted recall notice apparently does not require U.S. hospitals necessarily to remove from use the ED-3490TK model — again, unless, for example, the duodenoscope (according to the terms of the company’s January field correction) shows “shows visible signs of wear or physical damage.” According to the recall notice, 519 ED-3490TK devices sold in the U.S. were affected.
The mcr-1 gene
The mcr-1 gene has only recently emerged, according to a report the federal CDC published last year. This gene was first reported only approximately two years ago, in 2015, in food, animal, and patient isolates from China “and is notable for being the first plasmid-mediated colistin resistance mechanism to be identified.”
Pending additional data and information, the regulatory report Pentax filed in August would appear to indicate that endoscopy (namely, ERCP) may now be an emerging risk factor for infections caused by superbugs that can carry the mcr-1 gene.
An article published this year by the University of Minnesota’s Center for Infectious Disease Research and Policy concludes that: “Among the most worrisome scenarios is the emergence of bacteria that harbors the mcr-1 gene along with other antibiotic-resistance genes.”
Also according to this CIDRP article, as of last January, “MCR-1 has been detected in five human isolates in the United States. The US Centers for Disease Control and Prevention says it has increased surveillance for the gene in healthcare settings.” The incident described in Pentax’s August regulatory report might require that CIDRP’s numbers be updated. (Whether the CDC will publish a “notes from the field” report discussing the findings in this August regulatory report is unclear.)
Ominously, the CIDRP’s article warned further that, “If (CRE) were to acquire the (mcr-1) gene, for example, it could present clinicians with infections that are nearly impossible to treat with current antibiotics.”
Technical Consultant: Dr. Muscarella provides expert opinions, case reviews and advice for hospitals, device manufacturers and consumers about the causes of healthcare-associated infections (“HAIs”) linked to contaminated upper and lower GI endoscopes, among other devices. Contact him (using your smart phone): Email | Text (SMS) | Skype
More medical device reports
Pentax has filed other regulatory reports with the FDA this year, too, including several reporting that inspected ED-3490TK duodenoscopes failed to meet certain quality criteria, which the company had previously established. A significant number of these filed reports are similar in both content and format.
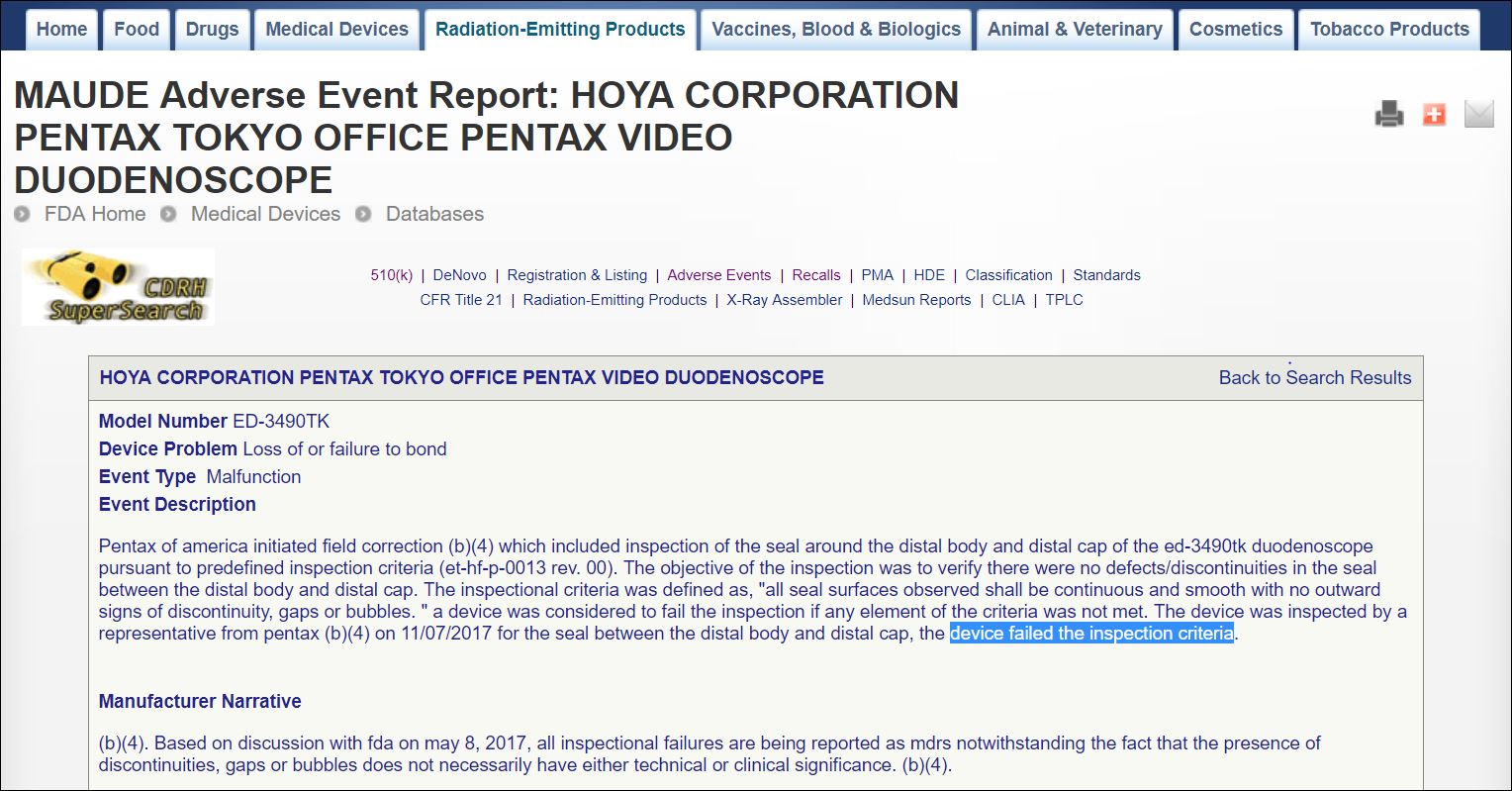
One of these reports [see inserted image, right] filed two weeks ago on November 29th, states that — pursuant to the terms of the company’s both January 16th (2017) field correction letter and July 11th (2017) recall, and, too, as discussed in the FDA’s January (2017) safety communication — a manufacturer representative inspected a ED-3490TK device on November 7th (2017) “to verify there were no defects/discontinuities in the seal between the distal body and distal cap.”
The November report states, however, that “the device was inspected by a representative from (P)entax [REDACTED] on 11/07/2017 for the seal between the distal body and distal cap” and that “the device failed the inspection criteria.”
As noted in the inserted image, above right, the company’s inspectional criteria was defined as “‘all seal surfaces observed shall be continuous and smooth with no outward signs of discontinuity, gaps or bubbles.’ A device was considered to fail the inspection if any element of the criteria was not met.”
For its part, Pentax states in this report that “the presence of discontinuities, gaps or bubbles (identified in an inspected ED-3490TK device) does not necessarily have either technical or clinical significance.”
Noted in the regulatory report the company filed in August, Pentax reported that the ED-3490TK device involved in this case of possible transmission of a colistin-resistant superbug during ERCP was “manufactured under normal conditions, passed all required inspections, and was released accordingly,” and had not been “reworked.”
Pentax’s finding would seem to confirm that the company had previously inspected this specific duodenoscope and did not identify any quality issues, such as cracks or gaps in the device’s adhesive, pursuant to the terms of its January field correction letter.
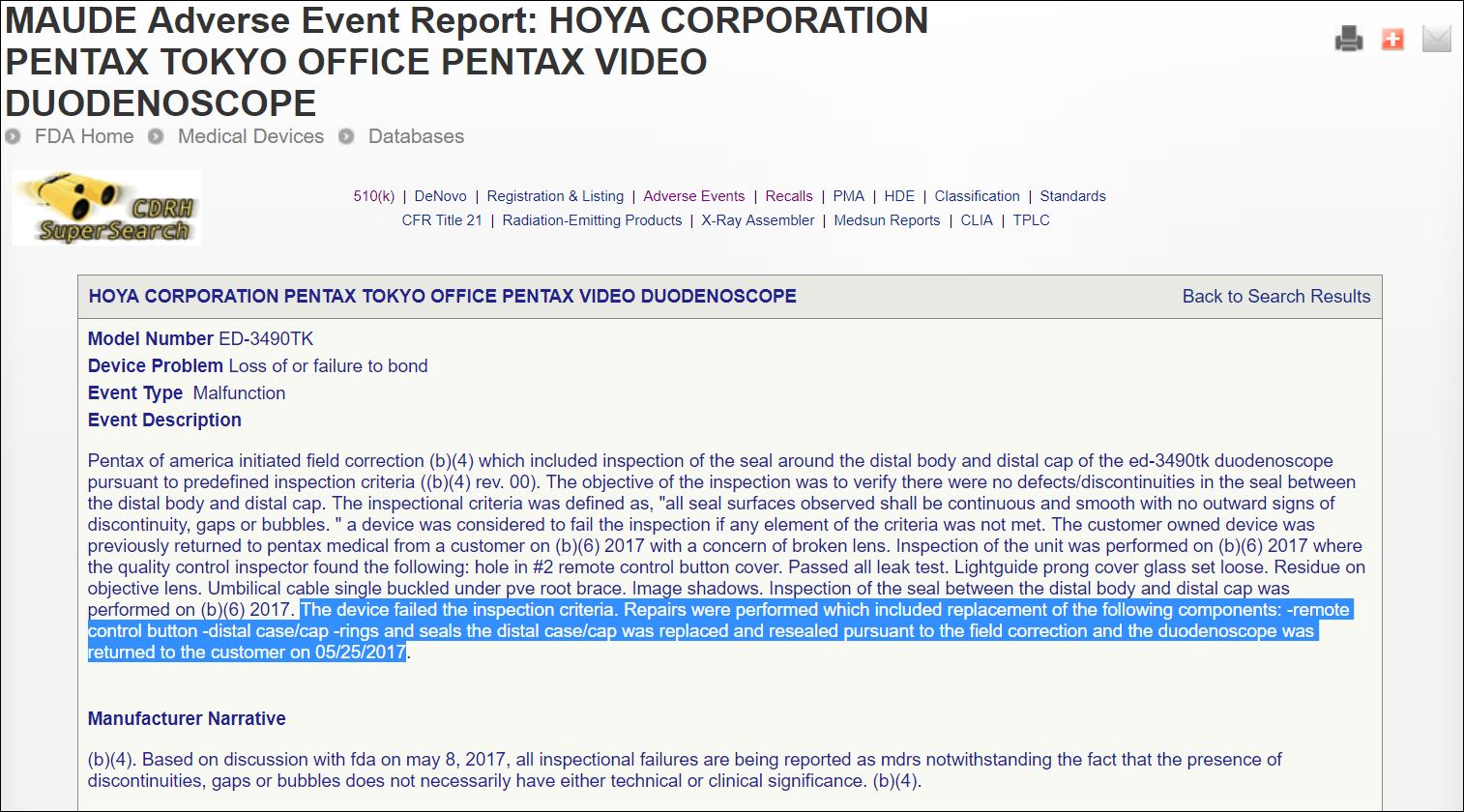
Another similar regulatory report the manufacturer filed with the FDA last spring, on June 20th, is provided in the inserted image, right.
Studies identifying the quality failures that the manufacturer found during the inspections of ED-3490TK duodenoscopes (as described in these filed regulatory reports) to be the direct cause of any one specific incident, infection or bacterial outbreak have not been published.
This June 20th regulatory report states that [see insert image, above], ” … the distal case/cap was replaced and resealed pursuant to the field correction and the duodenoscope was returned to the customer on (May 25, 2017).”
The ED-3490TK duodenoscope model was cleared by the FDA in 2009. The FDA cleared two similar duodenoscope models with a sealed design — the Olympus TJF-Q180V, last year — and the FujiFilm ED-530XT, in July. (All three models have been linked in the U.S. to CRE infections as discussed in detail in the aforementioned 2016 U.S. congressional report.)
In January 2016, when the FDA cleared the Olympus TJF-Q180V model, the FDA announced that, “We have made it a top priority to improve the safety of duodenoscopes and help protect patients from bacterial infections associated with these medical devices. … The Olympus TJF-Q180V’s new design, as well as the new annual inspection program, is intended to reduce the risk of fluid leakage into the elevator channel, which in turn can reduce patient exposure to bacteria and other potential infections.”
In September, Pentax received clearance from the FDA to market a novel duodenoscope with a disposable distal cap.
Article by: Lawrence F Muscarella, PhD. Posted: Dec. 8, 2017; updated Dec. 12. LFM Healthcare Solutions, LLC. Copyright 2017. LFM Healthcare Solutions, LLC. All rights reserved. V5
Lawrence F Muscarella PhD is the owner of LFM Healthcare Solutions, LLC, a Pennsylvania-based quality improvement and consulting company that provides safety services for hospitals, manufacturers and the public. Email Dr. Muscarella for more details.